Методика приготовления активированных окисленных (модифицированных) углей марки БАУ в различных ионных формах и их применение.
Очистка ионным обменом на катионитах и анионитах по сей день является значимым и распространённым методом для разделения и концентрирования элементов, глубокого извлечения примесей и с каждым годом подобные методики применяется всё шире, в том числе и для получения органических и неорганических реактивов напрямую.
При очистке окисленными углями в ионной форме нужно учитывать чистоту исходного реактива, рН среду, время контакта угля с раствором при статическом внесении или скорость пропускания раствора через угольную колонку. В случае статического внесения угля равновесная концентрация в среднем устанавливается максимум за 24 часа. Срок установления равновесия между фазами зависит от многих факторов. Для одних процессов обмена требуется до нескольких суток. В некоторых случаях ионный обмен проходит за меньший срок 5-6 часов. Это зависит от среды раствора; дисперсности или величины гранул угля; степени и способа его окисления; его плотности и проницаемости для обменного процесса; наличие и устойчивости различных комплексов металлов в растворе очищаемого реактива. Но эффективность и качество ионного обмена на углях всегда высокая.
Углями в ионных формах реактивы нужно только до очищать от следов примесей. Сильно загрязненные реактивы сначала очищают обычными методами согласно характеру находящихся примесей, методами, которые применяются для удаления основного их количества до квалификаций ч.д.а. и х.ч.
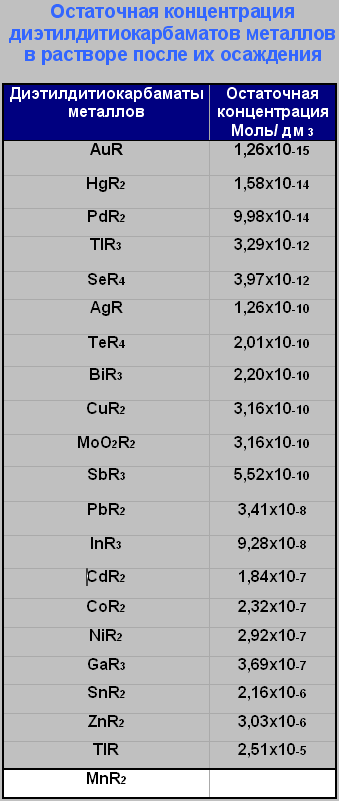
Для успешного использования углей в катионных формах по очистке какого либо реактива можно пользоваться избирательным рядом поглощения угля, а так же остаточной концентрацией комплексов металлов в растворе, после их осаждения. Приемлемая квалификация реактивов должна быть х.ч. или хотя бы ч.д.а, но с малыми количествами потенциальных примесных катионов для обмена, в суммарном количестве не более 0.001%.
Пользуясь избирательным рядом поглощения угля можно корректировать ионный состав среды очищаемого раствора для каждого отдельно взятого реактива, применив угли в соответствующих катионных формах. Если применяется уголь в Н+ форме для поглощения комплексов из очищаемого раствора, учитывают остаточную концентрацию комплексов и возможность замещения одного катиона металла другим в комплексе. Образцом может служить пример [9.б]. Если применяют уголь в форме какого либо металла, то руководствуются согласно ряду поглощения угля. Все данные приведены ниже.
Учитывая удельную емкость окисленного исходного угля в Н+ форме (приготовленного по способу см. HNO3 + карбамид), который, в отличие от других окисленных продуктов устойчиво ведёт себя как в слабокислой, так и в щелочной среде, на 1 литр 20% химически чистых растворов солей идет обычно от 5 до 10 г угля в катионной метало-форме, при условии, что реактив действительно соответствует своей присвоенной квалификации.
Для очистки растворов квалификации ч.д.а комплексно-адсорбционными методами количество угля в водородной форме в статическом внесении с запасом составляет от 10-15 г/л. Для реактивов квалификации х.ч. от 5 до 10 г/л. Для увеличения выхода очищаемого продукта и возможности сохранения постоянной рН растворов при этом способе применяют угли в катионной металлоформе.
Два ряда поглощения активного, модифицированного угля с уже исправленными неточностями, которые встречаются в первоисточниках:
1.
NH4+< Nа+< Cs+< Mg²+<Mn²+< Ca²+, Zn²+, Fe²+< Ni²+, Al³+< Cr³+< Cu²+< Fe³+
2.
NH4+< Nа+< K+< Rb+< Mg²+< Ca²+< Sr²+< Ba²+< Zn²+< Cd²+< Ni²+< Pb²+< Al³+< La³+< Cu²+< Fe³+
Пример 1. Щелочестойкий уголь в Н+ форме. Предварительно отмытый от золя, примесей и окисленный активированный уголь БАУ по методике HNO3 + карбамид, переводится 1-3% раствором соляной кислоты в Н+ форму. Контрольный образец - смесь угля и кислоты настаивают сутки, с контролем примесей тяжелых металлов колориметрически. При наличии примесей настаивание продолжают с новой порцией кислоты до отрицательного теста. Промывают водой, до рН 4 и сушат при 105°С. Применяют при средних значениях рН, для комплексной очистки реактивов, разделения и экстракции металлов с помощью комплексонатов, таких как 8-оксихинолин; диэтилдитиокарбамат натрия, калия или аммония; диметилглиоксим; рубеановодородная кислота и другие. Происходит обмен только по катиону с поглощением нерастворимых комплексов металлов.
Пример 2. Уголь в Na+ форме. Уголь в Н+ форме ( приготовленный по пункту 1 - здесь и далее) 20г помещают в фторопластовую или полиэтиленовую посуду и промывают 1-2% раствором очищенного NaOH с колориметрическим контролем на присутствие свободных гуматов (фульвокислот) в фильтрате. При их отсутствии уголь заливают свежим 1-2% раствором NaOH объёмом 200-250 мл или (предварительно очищенный сульфидами) 10%-ным раствором Na2CO3 с настоем смеси в течении суток и дальнейшей тщательной отмывкой от щёлочи. Применяют для коррекции ионного состава растворимых солей натрия и биологических жидкостей при рН 5-9 с настаиванием в течении 5-6 часов до суток*. Происходит только катионный обмен. Анионный состав раствора не имеет значения.
* В работах по измерению поглотительных свойств углей сроки обработки растворов углями в ионных формах иногда завышены намеренно или зависят от условий проводимых экспериментов. При большей временной обработке чем указано, есть возможность приготовления реактивов натрия спектрально чистых по каким либо катионам согласно ряду избирательности угля, при этом анализ показывает отсутствие спектральных линий примесных металлов.
Извлечение кальция и магния из городской водопроводной воды (при 20°С) до концентрации кальция 5,8-5,0х10-4% происходит за 4-5 часов.
Пример 3. Уголь в K+ форме. Уголь в Н+ форме помещают в фторопластовую или полиэтиленовую посуду и промывают 1-2% раствором очищенного КOH с колориметрическим контролем на присутствие свободных гуматов в фильтрате. При их отсутствии** уголь заливают свежим раствором КOH с настоем смеси в течении суток с дальнейшим полным отмыванием от щелочи. Применяют для коррекции ионного состава х.ч. растворимых солей калия и биологических жидкостей с одним или несколькими различными анионами в растворе при рН 5-9 с настаиванием в течении суток. Происходит обмен по катиону.
** При приготовлении различных катионных форм из одной партии окисленного угля анализ на свободные щелочерастворимые гуматы и при их отсутствии в дальнейшем не проводится.
Пример 4. Уголь в Са²+ форме. Уголь в Н+ форме 20 г заливают прозрачным, охлаждённым раствором очищенного и профильтрованного Са(OH)2, объемом 350 мл в пластиковой посуде, с настоем смеси в течении суток, при 0-10°С, с периодическим перемешиванием. Смесь изолируют от контакта с воздухом. После, уголь промывают водой не содержащей СО2 до нейтральной реакции промывных вод и сушат. Применяют для коррекции ионного состава растворимых солей кальция с одним или несколькими различными анионами в растворе при рН 5-9, с настаиванием в течении суток. Происходит обмен по катиону. Удаляются следы примесей Sr и Ba и тяжелых металлов согласно ряду поглощения угля. Большие количества изоморфных примесей удаляются не полностью, в связи с малой емкостью угля для изоморфов Са. В этом случае, обработку углем в Ca²+ форме повторяют, взяв его свежую порцию.
Пример 5. Уголь в Sr²+ форме. Готовится насыщенный водный раствор из очищенного Sr(ОН)2 при 80°С, фильтруется горячим и охлаждается до 10°С и снова фильтруется. Уголь БАУ 25 г заливают прозрачным раствором Sr(OH)2 объёмом 250-300 мл и настаивают 24 часа, затем отмывают водой. Применяется для коррекции ионного состава растворимых солей стронция при рН 5-9. На ряду с этим, возможна очистка двойных солей кальция и стронция - хлоридов сульфатов или ацетатов. Удаляются следы Ba и тяжелых металлов согласно ряду поглощения угля. Большие количества примеси Ва удаляются не полностью.
Перед применением уголя в Sr²+ форме, для удаления основного количества бария его осаждают в виде сульфата стандартной методикой.
Пример 6. Уголь в Ba²+форме. Уголь в Н+ форме обрабатывают аналогично пункту 5 раствором Ba(OH)2. Применяется для коррекции ионного состава растворимых солей бария. Удаляются следы тяжелых металлов согласно ряду поглощения угля.
Пример 7. Уголь в Mg²+ форме. Уголь в Н+ форме 20г помещают в полиэтиленовую, фторопластовую посуду и промывают 1-2% раствором очищенного NaOH или КОН согласно пункту 2, или берут готовый уголь в Na+ или K+ форме. (Уголь в NH4+ форме не применим). После промывки водой, полученный уголь в Na+ или К+ форме заливают 5%-ным раствором MgSO4, объёмом 200 мл, предварительно очищенный диэтилдитиокарбаматом натрия с поглощением выпавших комплексов металлов углем в Н+ форме. Смесь угля с раствором MgSO4, с установленной заранее нейтральной рН настаивают при комнатной температуре в течении суток, с периодическим перемешиванием, затем сливают раствор и заливают свежим раствором сульфата магния. После настаивания с перемешиванием в течении 5-6 часов, уголь моют водой от сульфат иона и сушат. Таким образом, путем катионного обмена, при дробной обработке получают качественный уголь в Mg²+ форме, применяемый для очистки растворимых солей магния и биологических жидкостей, где присутствие сторонних органических примесей, таких, как следов комлексообразователей или примесей ионов щелочных металлов не допускается. Очистку ведут при рН 5.5-8.
Пример 8. Уголь в NH4+ форме. Высушенный уголь в Н+ форме помещают в фторопластовую или полиэтиленовую посуду и промывают 5-8 % раствором особо чистого аммиака, с контролем за свободными гуматами***. После анализа основное количество угля заливают свежим раствором в концентрации 1-2% и настаивают смесь 6 часов в закрытой посуде. После, уголь моют водой до отсутствия запаха аммиака и высушивают при 105°С. Уголь является щелочестойким. Применяется для очистки всех растворимых солей, где аммиак в соединении выступает в качестве катиона. Очистка двойных и тройных солей аммония, в паре с катионами каких либо металлов, таким углём в NH4+ форме не допускается. Возможна очистка растворов карбоната аммония. Для разрушения комплексов меди, цинка, кадмия и других металлов в аммиачной среде, в растворы карбоната аммония вносят диэтилдитиокарбамат аммония или натрия, с последующим внесением угля в NH4 форме на 6-8 часов. Катионный обмен и ёмкость угля для щелочных металлов идет медленно, является величиной малой и при обычном количестве угля проходит не полностью. При необходимости обработку раствора реактива повторяют. Работы выполняются с обязательным контролем над примесями и использовании данных анализов.
*** Согласно прочим источникам щелочестойкий уголь в аммонийной форме приготавливают следующим способом. Окисленный уголь настаивают при перемешивании с 5-10%-ным раствором аммиака до образования стабильной и не изменяющейся желтой окраски раствора. Наиболее полное извлечение фульвокислот наступает после 1,5 - 2-х суток настаивания при периодическом перемешивании. Затем уголь моют дистиллированной водой до отсутствия запаха аммиака и сушат в шкафу при температуре до 100°С. Такой уголь больше не выделяет комплексообразующие фульвокислоты в любых щелочных средах.
Извлечение кальция и магния из водопроводной воды (при 20°С) до концентрации 5,0х10-4% происходит за 20-36 часов в зависимости от количества и качества угля.
Выдержка из работы студентов Новосибирский химико-технологического колледжа им. Д. И. Менделеева. 2003. Специальность 240138. Ивашева Д., Анаприленко Н.
"В работе использовался углекислый аммоний квалификации "х.ч.”, который содержал примеси по ГОСТ 3770-64. Тем не менее, после анализа реактива было установлено завышенное общее количество примесей металлов сероводородной группы, а количество железа и меди для полученного реактива составляло соответственно 0.0001% и 0.0003%. Очистку карбоната аммония проводили с использованием окисленного угля БАУ с отсеянным зерном менее 0,5 мм со статическим внесением в раствор. В качестве ориентира при поведении анализа служила примесь цинка, как наименее осаждаемый агент, имеющий в растворе аммиачный комплекс и примесь меди, как агент, наиболее осаждаемый для комплексообразователя диэтилдитиокарбамината аммония.
Реактив настаивали с углём в течение 6 часов. Количество примеси меди осталось в пределах исходного. Данный опыт указывает на невыполнимость очистки углями в катионных формах, в растворах при значениях рН начиная, примерно от 9 и выше. Катионный обмен оказался не эффективен и сходит на нет. Недостаточная очистка раствора карбоната аммония от следов меди без комплексообразователя в конкретном случае напрямую связана с высоким рН раствора и достаточной стойкостью амминокомплекса меди к катионному процессу обмена на углях в сильно щелочной среде. В связи с не разрушаемостью медно-аммиачного комплекса в щелочной среде катионным обменом с углями, в очищаемый раствор был предварительно внесён комплексообразователь в виде диэтилдитиокарбамината аммония, для разрушения медно-аммиачного комплекса и осаждения остальных примесей тяжёлых металлов в избыточном количестве равном 20% от необходимого. После разрушения аммиачного комплекса меди, при добавлении диэтилдитиокарбамината аммония и выпадении диэтилдитиокарбамината меди и прочих нерастворимых комплексов, уголь в аммонийной форме очистил раствор за 2 часа до низких, но не достаточных показателей содержания меди в растворе равным 2х10-5%.. Только после дальнейшего статического контакта раствора (NH4)2CO3 со свежей партией угля в течение 18 часов и указанного избытка комплексообразователя, анализ показал результат глубокого удаления примеси меди до остаточной концентрации равной 6х10-6%. При этом, количество примеси цинка снизилось с исходного 1х10-4% до конечного 4х10-5%. Наряду с этим, в растворе карбоната аммония наблюдается немногим меньшее извлечение примеси меди с использованием комплексообразователя, по сравнению с проведёнными аналогичными работами с применением растворов сульфата и хлорида аммония при нейтральных значениях рН.
Для дальнейшей очистки раствора карбоната аммония в раствор вносили перетёртый уголь БАУ в анионной форме для удаления избытка комплексообразователя, и после фильтрации реактив стандартно перекристаллизовали с предварительным насыщением раствора газообразным аммиаком и добавкой этанола для увеличения выхода. После перекристаллизации и промывки кристаллов ледяной водно-спиртовой смесью от маточного раствора препарат содержал по массе: Cu = 5х10-6%, Fe = 8х10-6%, Zn = 4х10-5%.
Из этих данных анализа следует, что ощутимых результатов по очистке карбоната аммония стандартной перекристаллизацией для эффективного удаления следов примесей амино-комплексов не наблюдается.
Вывод. Работа по очистке растворов карбоната аммония комплексно-адсорбционными методами показала эффективность и глубокое удаление растворённых примесей железа, а так же меди и цинка, имеющих устойчивые аммиачные комплексы в сильно щёлочных растворах по сравнению с стандартными методами. При этом для разрушения аммиачных комплексов был применён групповой комплексообразователь диэтилдитиокарбаминат аммония с последующим его выведением из раствора окисленными углями. Данная комплексно-адсорбционная методика дает возможность готовить препараты высокой чистоты и наиболее полно удалять сопутствующие примеси металлов в случаях очистки технических продуктов и соединений реактивных квалификаций, или когда методы синтеза из особо чистых веществ не применимы или не могут быть выполнены по каким либо причинам".
Пример 9. Уголь в Zn²+ форме.
а) В разбавленный в 2-3%-ный раствор соли цинка хлорида, ацетата или сульфата, предварительно очищенных от марганца, вносят 3% раствор диэтилдитиокарбамата натрия для осаждения части диэтилдитиокарбамата цинка - около 0,5-0,6% от всего количества цинковой соли. Смесь кипятят 10 минут для реакции избытка белого Zn-ДЭДК с примесными катионами металлов в растворах и их замещения в комплексе. (См. равновесные концентрации диэтилдитиокарбаматов металлов). Эквивалентная часть цинка, при этом, переходит в раствор, а цвет осадка комплекса меняет свой цвет. После кипячения раствор остужают до комнатной температуры и вносят уголь в Н+ форме на пол часа. Затем раствор хорошо фильтруют от угля, фильтрат разбавляют водой в два раза и снова приливают Na-ДЭДК, в количестве, для не полного осаждения цинкового комплекса. После перемешивания снова вносят уголь в Н+ форме на пол часа. На этот раз, уголь с поглощённым цинковым комплексом отфильтровывают от раствора, тщательно промывают водой со взбалтыванием и до удаления сторонних ионов хлора, ацетата или сульфата. Уголь в цинковой форме диэтилдитиокарбамата сушат около 105°С. Получают уголь с тёмно-серой, матовой поверхностью без блеска, для очистки солей цинка с возможностью обмена катионов примесей из раствора на ионы цинка в комплексе. Обмен только по катиону. Анионный состав для угля в Zn²+ форме не имеет значения. Очистку солей цинка с углем в Zn-ДЭДК форме проводят с одновременным внесением меньшего количества угля в H+ форме в соотношении (Zn-ДЭДК // H+) = 5:1. Дополнительно, таким углем можно проводить обмен по катионам в растворах двойных средних солей цинка-аммония.
б)**** Уголь для тонкой очистки растворов солей цинка получают следующим образом. Уголь в Н+ форме помещают в фторопластовую или полиэтиленовую посуду и промывают 1-2% раствором очищенного NaOH или КОН согласно пункту 2, или берут готовый уголь в Na+ или K+ форме. После тщательной промывки от щелочей, полученный уголь при 20-40°С, заливают прозрачным 5-10% раствором сульфата цинка у которого заранее установлена максимальная рН, но без допуска выпадения основных солей цинка при 20°С и выдерживают при периодическом перемешивании 24 часа при комнатной температуре*****. После проведения обмена и отмывки от сульфат иона, получаем уголь в Zn²+ форме, пригодный для очистки любых растворимых солей цинка, при средних значениях рН, от примесных следов кадмия, а так же прочих металлов, (см. ряд поглощения угля). Примеси щелочноземельных катионов и Mn²+ остаются в растворе после обмена. Дополнительно, таким углем можно проводить обмен по катионам в растворах двойных средних солей цинка-аммония.
**** Этот способ дает возможность приготовление угля с меньшей ёмкостью - примерно в половину или 1/3 от емкости угля приготовленного по пункту [а].
***** Для установления полного равновесия требуется около 2 суток.
Пример 10. Уголь в Li+ форме. Уголь в Н+ форме обрабатывают аналогично пункту 2 растворами 1%-ным LiOH или насыщенным горячим Li2CO3 с последующим остыванием и настаиванием в течении 24 часов. Промывку от карбоната ведут горячей водой. Применяют при доочистке каких либо литиевых растворимых солей и технических жидкостей электролитов при нейтральных и слабо щелочных значениях рН.
Регенерация всех катионных форм угля производится обратным переводом в Н+ форму. Проводится промывка 5-10% раствором сначала х.ч., затем ос.ч. соляной кислоты с настоем или кипячением, и промежуточными промывками водой между промывками кислотой, до тех пор, пока анализ промывной кислой жидкости не будет показывать на отсутствие сторонних ионов. В зависимости от величины гранул и проницаемости угольного материала, регенерация может занять время до нескольких суток. Быстрая и скоротечная промывка кислотой крупно фракционного угля БАУ обычно не приводит к возможно полному удалению накопленных примесей из-за недостаточного извлечения последних из глубинных слоёв угля.
Заводская Лаборатория.
Нужно учитывать, что катионный обмен в слабокислой среде в районе рН 4.5-4 и менее, на углях в металлоформе проходит не эффективно. Для проведения работ с успешным результатом, рН растворов устанавливается в районе 6.5-8.5, с учетом использования щелочестойкого окисленного угольного материала. При использовании угля не стойкого к щелочам рН растворов уменьшают до 8.
Все работы проводятся с постоянным контролем и использованием результатов анализа.
С Ув. Argentus/2011
|  ПРИГОТОВЛЕНИЕ ОКИСЛЕННОГО УГЛЯ МАРКИ БАУ В РАЗЛИЧНЫХ ИОННЫХ ФОРМАХ
ПРИГОТОВЛЕНИЕ ОКИСЛЕННОГО УГЛЯ МАРКИ БАУ В РАЗЛИЧНЫХ ИОННЫХ ФОРМАХ